| You
are here: Home: BCU 1|2003: Dr.
Larry Norton, MD
 |
|
|
|
 |
Larry
Norton, MD
|
|
|
Attending Physician and Member,
Memorial Hospital
Head, Solid Tumor Division,
Norna S Sarofim Chair in Clinical Oncology
Medical Director,
Memorial Sloan-Kettering Cancer Center
Chairman, Medical Advisory Board,
Breast Cancer Research Foundation
Immediate Past President,
American Society of Clinical Oncology
Chair, Breast Committee,
Cancer and Leukemia Group B |
|
 |
Edited comments by Dr Norton
Evolution of the Norton-Simon Hypothesis
The study design of CALGB 9741 was based on rigorous mathematical
modeling, which generated clinical trial data and then generated
this experiment. This study had a 25-year history, starting with
a clinical observation, which led to a theory, which led to experiments
to refine the model, which generated new experiments and eventually
led to these results.
The original clinical observation was a patient I saw with Hodgkin’s
disease when I was a clinical associate at the National Cancer Institute.
He finished six cycles of MOPP chemotherapy and did very well for
over a year, but relapsed in the same sites with the exact same
histology approximately 17 months later. We put him back on MOPP
chemotherapy, and again he had a spectacular response. We saw growth,
regression and regrowth.
My background was in mathematics, and I worked with Richard Simon
from the National Cancer Institute to graph this out and try to
fit curves to it. But when we tried to fit the existing models to
the data on this particular patient, it just didn’t work.
Mathematical models for tumor growth
We looked at the Skipper-Schabel model, which says that exponential
growth is constant log growth, and exponential regression translates
to constant log kill. If a tumor doubles in a certain period of
time, it will double in that period of time no matter how big it
is. If it shrinks by half over a period of time in response to therapy,
it will always shrink by half. Gompertzian growth is exponential
growth with a constant exponential regression.
The question was how the Skipper-Schabel model applied to Gompertzian
growth. It was very clear looking at this patient’s record
that his response was not log kill. It turned out to be simple —
if the tumor grew in a Gompertzian fashion, it would regress in
a Gompertzian fashion.
When I graphed it, this patient fit so perfectly that I could
accurately predict when he would go in complete remission. As long
as you have homogeneity in response to therapy, the model worked
very well. This led to a series of laboratory experiments and clinical
trials. Using this model, deviations from Gompertzian growth are
due to drug resistance — the emergence of different clones
with different growth kinetics and responses to therapy.
Dose-dense therapy targets inhibition of regrowth
Apaper in Seminars in Oncology in the mid-1980s indicated that
the primary problem in Gompertzian growth is not cell kill, but
rather regrowth between cycles. While therapy gets us closer to
the cure limits, you have to get below a small number of cells to
prevent regrowth, and you regrow faster away from that limit. There's
a rebound effect, and the key is to inhibit that regrowth.
One of the simplest ways to address regrowth is to move the doses
of therapy close enough together to have less regrowth between cycles.
This is extremely powerful in Gompertzian kinetics, as long as you
can drive the tumor toward that cure limit. In the adjuvant setting,
when you’re probably close to the cure limit, you can have
dramatic benefits by giving the doses closer together in time.
| The
Gompertzian model and tumor regrowth |
"In
the Gompertzian model, smaller tumors grow faster, so tumor
regrowth between treatment cycles is more rapid when cell
kill is greatest. Reducing the time available for tumor regrowth
(increasing dose density), which is now possible through the
use of colony-stimulating factors to hasten hematopoietic
recovery, may have a greater impact on clinical outcome than
dose escalation. Sequential schedules allow optimal doses
to be used in dose-dense cycles." |
| DERIVED
FROM: Norton L. Evolving concepts
in the systemic drug therapy of breast cancer. Semin Onc 1997;24(4
Suppl 10):S10-3-S10-10. Abstract. |
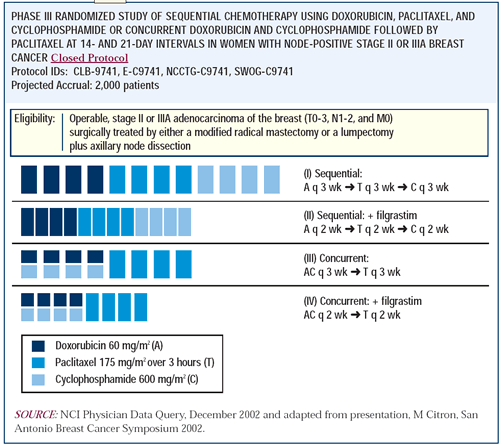
CALGB 9741: Phase III study of dose-dense and
sequential adjuvant chemotherapy
This study was designed with input from all members of the breast
Intergroup and coordinated by the CALGB. It had a two-by-two factorial
design. The two parameters were dose-density — giving drugs
every two weeks instead of every three weeks using G-CSF —
and combination versus sequential therapy. The doses were the same
optimal doses derived from previous clinical trial experience. The
only difference was the schedules.
Improved survival with less toxicity with dose-dense
chemotherapy
The study demonstrates a considerable advantage to dose density
in diseasefree survival — the primary endpoint of the study
— and overall survival. There was an approximate 31 percent
reduction in the annual odds of death with the dose-dense therapy.
This benefit was not at the cost of increased toxicity. In fact,
the dose-dense regimens were less toxic than the conventional regimens,
particularly in terms of neutropenia. In every important parameter
except for anemia, dose-dense therapy was superior in terms of toxicity.
Sequential dose-dense therapy eliminated the anemia while maintaining
preservation of efficacy.
This is one of the first regimens I’ve seen where there’s
nothing "not to love." It’s more efficacious, less
toxic and over more quickly. The incidence of longer-term effects,
so far, is the same as we would expect from the drugs without dose
density. In retrospect, it is logical — you’re giving
G-CSF for neutropenia, getting the drugs in more quickly, leaving
less time to develop other toxicities and obtaining more efficacy
because more drug is given over a shorter period of time.

The importance of hematopoetic support during
dose-dense therapy
One key factor in the dose-dense approach is the use of granulocyte
colonystimulating factors. Some form of granulocyte stimulation
is absolutely essential, and this trial utilized filgrastim on days
3-10.
Monica Fornier, Cliff Hudis and colleagues are planning a study
to look at the longer-acting formulation — pegfilgrastim.
We have every reason to believe this agent will be both very effective
and more convenient for the patient. Once we have the feasibility
data, which should be fairly soon, I think the longer-acting formulation
could be utilized for regimens like this.
Clinical applicability of dose-dense adjuvant
chemotherapy
Dose-dense adjuvant chemotherapy in a nonprotocol setting is a
reasonable option. This trial, which accrued over 2,000 patients,
shows improved efficacy, decreased death rates and reduced toxicity;
therefore, there’s no reason not to use dose-dense therapy
at this time.
I believe in dose-dense therapy because I’ve seen its evolution
in the laboratory and the clinic for 25 years, and I believe it
has a solid basis. However, no individual can stand up and say this
is the new standard of care. We have to see how people are going
to utilize this in the community. I would not be shocked to find
this approach widely accepted and used, but whether it becomes a
new standard of care needs to be defined by the community.
Optimal dosing and scheduling in dose-dense chemotherapy
I am concerned physicians will be enthralled with the idea of
giving the doses closer together and reduce the doses in order to
accomplish that. This may or may not work, depending on the cell
kill per dose. I would rather use 60 mg/m2 of doxorubicin as often
as possible than go down to 30 or 40 mg/m2 of doxorubicin to give
it more often.
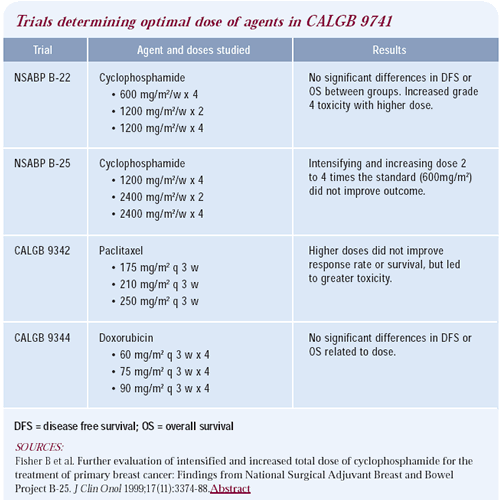
The doses selected for this trial were based on previous studies.
CALGB 9344 demonstrated that doses greater than 60 mg/m2 of doxorubicin
do not convey any advantage, and that the addition of paclitaxel
made an important difference, especially in the subset of patients
with estrogen receptor-negative tumors.
Another CALGB study of patients with stage IV advanced disease
by Eric Winer showed that doses of paclitaxel higher than 175 mg/m2
were associated with more toxicity and no significant advantage.
The NSABP also did a series of excellent randomized trials, which
showed that the efficacy of cyclophosphamide was capped at the dose
of 600 mg/m2.
We chose the every two-week schedule for trial CALGB 9741 out of
convenience. There's no real reason it has to be two weeks. In fact,
with G-CSF, most patients are ready to be treated after 10 or 11
days. If changing from 21 days to 14 days results in a one-third
reduction in mortality, then going from 14 days to 10 days may result
in a further reduction in mortality. These are the kinds of regimens
that we need to start testing prospectively in clinical trials.
Dose and schedule of adjuvant paclitaxel
The issue of weekly dosing of taxanes — specifically paclitaxel
— is an open question. Weekly administration certainly reduces
toxicity and seems to preserve response rate. The CALGB is accruing
patients to a study comparing weekly versus every three-week paclitaxel.
This trial is also asking questions about trastuzumab use in the
setting of metastatic breast cancer. While this is a very important
trial, the weekly paclitaxel is being given at a compromised dose.
It uses 80 mg/m2 per week, which adds up to a high cumulative dose,
but we don’t know the dose-response curve for paclitaxel at
that level. It’s possible that 175 mg/m2 every two weeks is
more effective in cell kill than 80 mg/m2 every week.
You can give more than 80 mg/m2 per week of paclitaxel for a few
cycles, but then you see significant toxicity, particularly neurotoxicity.
There are interesting agents being investigated in terms of their
ability to ameliorate the neurotoxicity of paclitaxel enough to
give a higher dose every week. It may even be possible to approach
the every two-week dose on a weekly basis by using G-CSF.

Trastuzumab as first-line therapy in metastatic
disease
The pivotal trial of trastuzumab clearly showed a survival advantage
to trastuzumab combined with chemotherapy — either AC or paclitaxel.
AC/trastuzumab led to a higher-than-expected — and higher-thanacceptable
— incidence of cardiotoxicity, so doxorubicin is not widely
used with trastuzumab. Paclitaxel with trastuzumab in the HER2-positive
situation clearly results in a survival advantage, and it does not
make sense to deny patients that survival advantage.
We still don’t know how long to continue trastuzumab after
disease progression, and there is a current MD Anderson trial evaluating
this. The clinical trial community needs to address this issue.
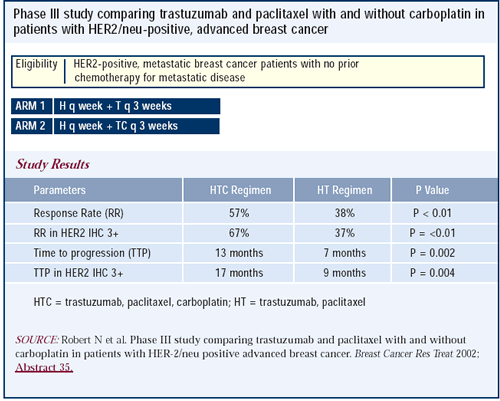
Phase III trial of trastuzumab and paclitaxel
with or without carboplatin in advanced breast cancer
Platinums are active agents, and there is evidence of benefit
in combining them with trastuzumab. Nicholas Robert is reporting
an important phase III study comparing trastuzumab and paclitaxel
with and without carboplatin in patients with HER2-positive advanced
breast cancer. The addition of carboplatin increased the response
rate and the duration of response. I think it is important to find
out whether these agents need to be combined to obtain the desired
result, or whether they can be given sequentially.
Advantages of capecitabine in the management of
metastatic disease
Capecitabine is an excellent agent. From a cell kinetics perspective,
it achieves very high intracellular levels of 5-fluorouracil, so
dose is not compromised. Oral administration is a significant advantage.
Patients with disease progression on hormonal therapy who are not
psychologically ready for intravenous therapy can go from their
hormone pill to capecitabine without a big transition. It is oral
and can be administered frequently, giving a high-dose bolus of
5-fluorouracil. Patients do not have a lot of toxicity if the dose
is monitored carefully.
We give capecitabine two weeks on followed by one week off, but
this may not be the optimal schedule. Many of us have talked for
years about exploring other schedules of administration, and I would
like to see more innovative schedules of capecitabine tested in
clinical trials. A higher dose given weekly or every five or ten
days would give the patient a very high dose of 5-fluorouracil.
I'm not sure we have achieved the optimal schedule for this important
active agent.
Capecitabine plus docetaxel as combination therapy
Capecitabine and docetaxel are both very active agents. In advanced
disease, when we’re dealing with heterogeneous drug sensitivity,
studying the drugs together makes a lot of sense. A higher percentage
of patients may respond to therapy with improved duration of disease
control because some patients will benefit who wouldn’t have
before.
I’ve treated some patients with stage IV disease who responded
brilliantly to docetaxel but did not respond well to capecitabine.
Others respond to capecitabine, but not to docetaxel.
If a given patient has a largely capecitabine-sensitive tumor,
I’d be better off treating with capecitabine than with the
capecitabine/docetaxel combination. If response is my primary goal,
and I don't know whether the tumor is responsive to capecitabine,
I'm better off giving the combination more.
However, I don’t know what happens if I treat with docetaxel,
and switch to capecitabine if I don’t see an early response.
This is somewhat different from the common clinical practice of
treating for a long time before switching. I think we wait too long
before changing therapies in general, because we actually wait for
tumor regrowth. Even if we obtain a good response to one drug, we
wait for tumor regrowth before we switch, and we probably should
do this sooner.
I think this is one of the key things we have to start looking
at in terms of clinical trials and monitoring patients. PET scanning
and tumor markers might be very useful in this regard. It might
be more advantageous to change therapies when the tumor markers
rise than when there’s imaging evidence of disease progression.
Earlier diagnosis of metastatic disease may make a significant
difference if your therapy is effective. At Memorial Sloan-Kettering
Cancer Center we have a tumor vaccine protocol for patients with
rising markers without clinical evidence of disease. This is where
vaccines may make a difference.
Select publications
|