|
||||||||

| Tracks 1-17 | ||||||||||||||||||||||||||||||||||||
|
Select Excerpts from the Interview
Tracks 2-4
![]() DR LOVE: Can you discuss the Gompertzian growth hypothesis and how
it relates to dose-dense chemotherapy?
DR LOVE: Can you discuss the Gompertzian growth hypothesis and how
it relates to dose-dense chemotherapy?
![]() DR NORTON: We’ve been interested in Gompertzian growth for decades,
which in addition to being interesting has clinical applications. The whole
concept of dose density or the scheduling of chemotherapy drugs being at least
as important as dose level is dependent on the fact that tumors grow in this
Gompertzian fashion.
DR NORTON: We’ve been interested in Gompertzian growth for decades,
which in addition to being interesting has clinical applications. The whole
concept of dose density or the scheduling of chemotherapy drugs being at least
as important as dose level is dependent on the fact that tumors grow in this
Gompertzian fashion.
They start to grow exponentially, but growth eventually trails off and reaches a plateau phase.
No counterexamples have emerged, and everything that’s ever been observed to grow in nature, including cancer tumors, follows this kind of growth pattern. Why things follow this growth pattern or why it’s so ubiquitous in nature has never been clear.
Understanding why, obviously, could be of tremendous importance because it would not only explain why dose density works, which is derived from the mathematics of Gompertzian growth, but it would also potentially provide some therapeutic targets.
![]() DR LOVE: Can you discuss your self-seeding hypothesis (Norton 2006)?
DR LOVE: Can you discuss your self-seeding hypothesis (Norton 2006)?
![]() DR NORTON: I received a phone call from one of my collaborators, Joan
Massagué, who has been studying metastasis. He took human breast cancer
cells, grew them in appropriate animal models and found cells that were
metastasizing to the lung. He then took those cells, reimplanted them in the
mammary fat pad of the recipient mice, and developed cell lines that had a
high potential for lung metastasis (Minn 2005).
DR NORTON: I received a phone call from one of my collaborators, Joan
Massagué, who has been studying metastasis. He took human breast cancer
cells, grew them in appropriate animal models and found cells that were
metastasizing to the lung. He then took those cells, reimplanted them in the
mammary fat pad of the recipient mice, and developed cell lines that had a
high potential for lung metastasis (Minn 2005).
The cell lines with a high probability of metastasizing to the lung grew very rapidly in the mammary fat pad, the primary site in which they were implanted. Also, the genes that were overexpressed in the cell lines with potential for lung metastasis — compared to the genes in the parental cell line — were not associated with cell division, apoptosis or cell loss. They were the genes associated with matrix dissolution, angiogenesis, cell adhesion, et cetera (Minn 2005).
In that discussion, it became evident that one way of explaining the phenomenon of the tumor growing faster in the mammary fat pad was not that the cells were dividing more rapidly but that they were metastasizing back to themselves. In other words, the cells leave the mammary fat pad, and some go to the lung, but some go into circulation and come back to the tumor where they originated.
It’s logical from a biologic point of view because that is the organ they’re most comfortable in and that is where they were growing in the first place. It would also explain a lot about cancer that, right now, is mysterious. Why is cancer so disorganized histologically? Maybe it’s disorganized because tumors are not one entity but a collection of little entities (ie, little metastases). Maybe they’re growing quickly because a tumor is not one big mass but a collection of little masses.
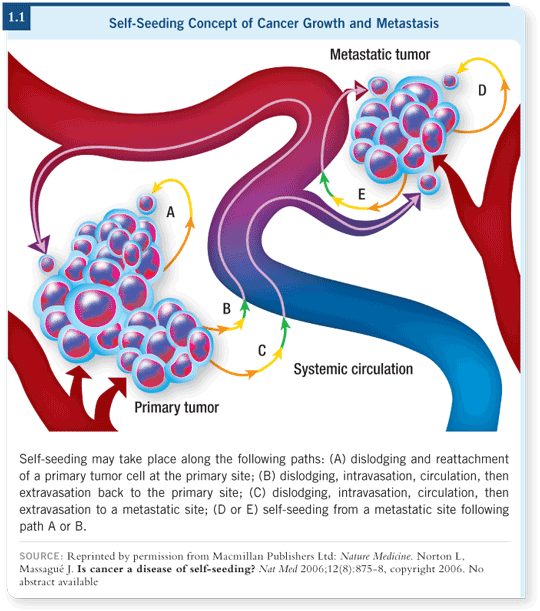
The bottom line is that we’re convinced aggressive cancers attract their own cells that go out into circulation. We’ve termed this “self-seeding” because it’s reminiscent of the way weeds take over your garden (Norton 2006; [1.1]).
A weed takes over your garden by seeding other weed plants. Each weed plant is not particularly fast growing or large. It’s not the weed that takes over your garden — it’s the weed bed. Weeds are invasive — they invade the normal plants in your garden — and they are metastatic because the same seeds that can fall in your garden can fall in your neighbor’s garden.
It ties together closely with the stem cell theory because the seeds may indeed be stem cells. That’s why they are causing continued growth wherever they’re found, because each one is a nidus of another tumor focus.
Tracks 5-7
![]() DR LOVE: Can you discuss the clinical and therapeutic relevance of this
hypothesis?
DR LOVE: Can you discuss the clinical and therapeutic relevance of this
hypothesis?
![]() DR NORTON: From a therapeutic perspective, the self-seeding concept is fascinating because we have no drugs to interfere with that process. However,
that process should be a rich source of targets because the ability to migrate
through the tissues, break away from the primary tumor mass, go into a blood
vessel, travel and survive in the blood vessel, come out of the blood vessel,
reanchor itself and start to divide involves molecules that must be located on
the surface of the cell.
DR NORTON: From a therapeutic perspective, the self-seeding concept is fascinating because we have no drugs to interfere with that process. However,
that process should be a rich source of targets because the ability to migrate
through the tissues, break away from the primary tumor mass, go into a blood
vessel, travel and survive in the blood vessel, come out of the blood vessel,
reanchor itself and start to divide involves molecules that must be located on
the surface of the cell.
These are all cell-surface phenomena related to adhesion and trying to find a niche in which to grow and develop. It may be that antibodies or small molecules that work against cell-surface molecules are important. All of these are potential areas for drug development.
I can say for sure that I know the phenomenon occurs. How important it is and how it relates proportionally, in terms of malignancy, to other characteristics of cancer remains to be determined. Self-seeding, however, is a truth, and it is a potential target for intervention.
![]() DR LOVE: Are there agents that interfere with this cycle?
DR LOVE: Are there agents that interfere with this cycle?
![]() DR NORTON: Actually, we believe that the seeding of the metastasis is not
the primary problem. The primary problem is growth in metastatic sites.
If a “shower” of cancer cells occurred to somebody’s entire body and each
cell only divided two or three times to form a microscopic focus that never
became any larger, cancer would not be a problem.
DR NORTON: Actually, we believe that the seeding of the metastasis is not
the primary problem. The primary problem is growth in metastatic sites.
If a “shower” of cancer cells occurred to somebody’s entire body and each
cell only divided two or three times to form a microscopic focus that never
became any larger, cancer would not be a problem.
The important factor is the growth in the metastatic sites, and we have to consider the possibility that the growth in metastatic sites is also a result of self-seeding in the metastatic sites. The recent paper we published in Nature Medicine provides an illustration of this (Norton 2006; [1.1]).
This fascinates me as a biomathematician. If you hypothesize that self-seeding occurs, then self-seeding occurs from the outside in. In other words, cancer grows from the outside inward. The surface area of a mass is related to its diameter squared, whereas the volume of a mass is related to its diameter cubed. Because growth is occurring from the outside in, growth is proportional to the diameter squared, but you’re going to lose cells by spontaneous cell death related to the diameter cubed. The ratio of growth to death will drop over time, and that will give you the Gompertzian phenomenon.
If the hypothesis is true and the curve is an accurate representation of tumor growth, then we don’t have to kill cancer cells, necessarily, to be able to cure patients. We have to affect growth parameters so that each tumor doesn’t grow large — for example, increase cell death slightly or increase the spatial arrangement of the cells on the periphery of the tumor where the growth occurs.
I’m encouraged by the experts in cancer stem cells who have assured me they find cancer stem cells on the periphery of tumors, not in the core of tumors. If we can somehow make these stem cells deposit themselves in the tumor in a more diffuse pattern, rather than such a dense pattern, that may be enough to convert a malignant tumor to a benign mass.
Track 9
![]() DR LOVE: At another level, any speculation about how effective bevacizumab
will be in the adjuvant setting? How does that tie into the self-seeding
hypothesis?
DR LOVE: At another level, any speculation about how effective bevacizumab
will be in the adjuvant setting? How does that tie into the self-seeding
hypothesis?
![]() DR NORTON: Bevacizumab and other anti-angiogenic agents should be
studied in the adjuvant setting. In fact, if the self-seeding hypothesis is correct,
the earlier you use these agents the more effective they will be because the
metastatic process is dependent on seeding.
DR NORTON: Bevacizumab and other anti-angiogenic agents should be
studied in the adjuvant setting. In fact, if the self-seeding hypothesis is correct,
the earlier you use these agents the more effective they will be because the
metastatic process is dependent on seeding.
Early breast cancer, when it’s micrometastatic, might be the best time to intervene with the ability of seeds, which have already spread, to attract a blood supply. If anything, the self-seeding theory would suggest that anti-angiogenic therapy should be more active in the adjuvant setting than against advanced disease.
Track 14
![]() DR LOVE: Theoretically, how does dose-dense therapy tie into the self-seeding
hypothesis?
DR LOVE: Theoretically, how does dose-dense therapy tie into the self-seeding
hypothesis?

![]() DR NORTON: I believe it ties
into it because the concept
of dose density derives
mathematically from the
Gompertzian phenomenon.
Tumors grow and respond
to therapy in a Gompertzian
fashion — meaning if they
grow fast, they shrink fast
and if they grow slowly, they
shrink more slowly, which is the Norton-Simon hypothesis (1.2).
DR NORTON: I believe it ties
into it because the concept
of dose density derives
mathematically from the
Gompertzian phenomenon.
Tumors grow and respond
to therapy in a Gompertzian
fashion — meaning if they
grow fast, they shrink fast
and if they grow slowly, they
shrink more slowly, which is the Norton-Simon hypothesis (1.2).
If you plug into your thinking Gompertzian growth and regression proportional to rate of growth, then dose density stands out. It implies that the big problem is to kill cancer cells, but you have to come back in with another dose of therapy before they have a chance to regrow. So you pick the dose of the drug that provides the optimal response, and you administer it as often as you can.
Track 15
![]() DR LOVE: How does the concept of dose density relate to the work your
group at Memorial has been doing with capecitabine?
DR LOVE: How does the concept of dose density relate to the work your
group at Memorial has been doing with capecitabine?
![]() DR NORTON: We designed some experiments to observe the growth curves of
tumors in mice that were being treated with capecitabine. We found that six, seven, eight or nine days was the point at which the ratio of the regression to
its anticipated growth rate was maximum. If you kept treating at eight, nine,
10, 11, 12, 13 or 14 days, the tumor continued to shrink, but it was shrinking
more gradually than it was shrinking on day seven.
DR NORTON: We designed some experiments to observe the growth curves of
tumors in mice that were being treated with capecitabine. We found that six, seven, eight or nine days was the point at which the ratio of the regression to
its anticipated growth rate was maximum. If you kept treating at eight, nine,
10, 11, 12, 13 or 14 days, the tumor continued to shrink, but it was shrinking
more gradually than it was shrinking on day seven.
To optimize that schedule, one has to stop capecitabine at day seven and then come back with another seven days of therapy as soon as possible, which is the dose-dense concept. You pick the optimal dose and schedule and administer it as often as possible.
We explored, in animal models, capecitabine administered seven days on and seven days off. It is remarkable that we could drive the dose level much higher. If you don’t have to worry about the second week of therapy, you can push the dose level higher, which causes even more regression. We obtained fantastic results in the animal models.
Based on that evidence, Maria Theodoulou, Cliff Hudis and Tiffany Traina have been conducting at Memorial Sloan-Kettering a Phase I/II trial of capecitabine administered seven days on and seven days off. The trial is still ongoing because we can’t reach the maximum tolerated dose.
We’ve gone much higher with the capecitabine dose than we ever could have
imagined. Responses are terrific, and the toxicity is greatly minimized. Just as
we discovered with the use of filgrastim or pegfilgrastim with AC![]() paclitaxel
every two weeks (Citron 2003; Hudis 2005; Burstein 2005), we have greater
efficacy with less toxicity. The seven days on, seven days off with capecitabine
seems to be efficacious. In addition, it’s less toxic than 14 days on and seven
days off.
paclitaxel
every two weeks (Citron 2003; Hudis 2005; Burstein 2005), we have greater
efficacy with less toxicity. The seven days on, seven days off with capecitabine
seems to be efficacious. In addition, it’s less toxic than 14 days on and seven
days off.
![]() DR LOVE: Are we moving toward adjuvant dose-dense AC
DR LOVE: Are we moving toward adjuvant dose-dense AC![]() paclitaxel
followed by capecitabine?
paclitaxel
followed by capecitabine?
![]() DR NORTON: People are talking about that regimen now, especially in the
setting of preoperative dose-dense AC
DR NORTON: People are talking about that regimen now, especially in the
setting of preoperative dose-dense AC![]() T, because not all patients will have
a pathologic complete remission. If you use dose-dense AC
T, because not all patients will have
a pathologic complete remission. If you use dose-dense AC![]() T and you don’t
obtain a pathologic complete remission, then you have residual cells that are
probably resistant to those agents. Therefore, dose-dense capecitabine at that
point would be a reasonable idea. We’re also considering combinations of
dose-dense capecitabine with antivascular agents, such as bevacizumab, and
anti-HER2 agents.
T and you don’t
obtain a pathologic complete remission, then you have residual cells that are
probably resistant to those agents. Therefore, dose-dense capecitabine at that
point would be a reasonable idea. We’re also considering combinations of
dose-dense capecitabine with antivascular agents, such as bevacizumab, and
anti-HER2 agents.
Track 17
![]() DR LOVE: Where are we with dose-dense AC
DR LOVE: Where are we with dose-dense AC![]() paclitaxel/trastuzumab?
paclitaxel/trastuzumab?
![]() DR NORTON: Chau Dang, in our program, has completed a study, and we’re
still following the last patient out far enough to be able to publish definitive
results. We have, however, presented mature preliminary results several times
(Dang 2006, 2005).
DR NORTON: Chau Dang, in our program, has completed a study, and we’re
still following the last patient out far enough to be able to publish definitive
results. We have, however, presented mature preliminary results several times
(Dang 2006, 2005).
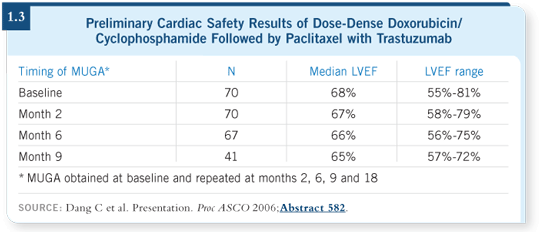
The major observation we’ve made is that cardiotoxicity is extremely acceptable (Dang 2006; [1.3]). We’re certainly not seeing any more cardiotoxicity with dose-dense AC paclitaxel/trastuzumab than with the nondose-dense use of those agents. We might be seeing less cardiotoxicity with dose-dense therapy than we saw before.
In CALGB-9741, dose-dense AC produced less cardiotoxicity than every three-week AC (Citron 2003; Hudis 2005). If the chemotherapy itself had less cardiotoxicity, then the additional incremental impact of trastuzumab would be less as well.
Currently, dose-dense AC![]() paclitaxel/trastuzumab is our standard of care for
adjuvant therapy in HER2-positive disease.
paclitaxel/trastuzumab is our standard of care for
adjuvant therapy in HER2-positive disease.