
|
||||||||

| Tracks 1-20 | ||||||||||||||||||||||||||||||||||||||||||
|
Select Excerpts from the Interview
Track 1
![]() DR LOVE: Can you discuss the background of your recently reported study of nab paclitaxel versus docetaxel?
DR LOVE: Can you discuss the background of your recently reported study of nab paclitaxel versus docetaxel?
![]() DR GRADISHAR: Nab paclitaxel was developed to take advantage of the significant
antitumor activity of the taxanes but also to avoid some of their side effects. Solvents typically used with drugs such as docetaxel or Cremophor®-
based paclitaxel are absent, and instead the paclitaxel is administered in an albumin delivery system to increase the amount of drug that reaches the tumor tissue. That’s the underlying rationale.
DR GRADISHAR: Nab paclitaxel was developed to take advantage of the significant
antitumor activity of the taxanes but also to avoid some of their side effects. Solvents typically used with drugs such as docetaxel or Cremophor®-
based paclitaxel are absent, and instead the paclitaxel is administered in an albumin delivery system to increase the amount of drug that reaches the tumor tissue. That’s the underlying rationale.
What’s been shown to date, both through some of the early Phase I/II trials and ultimately the Phase III trial, is that when administered every three weeks, nab paclitaxel was superior to solvent-based paclitaxel administered every three weeks (Gradishar 2005).
Despite more of the paclitaxel being administered in the nab preparation than with the every three-week solvent-based paclitaxel, less neutropenia occurred. A different kind of neuropathy appeared to be present that resolved more quickly. A greater antitumor effect was also observed in terms of response rate and improved progression-free survival.
In an era when we’re increasingly using weekly therapy and when many perceive docetaxel to be the most active single-agent anticancer therapy for breast cancer, what most people want to know is, how does nab paclitaxel compare to a weekly taxane schedule? How does it compare to docetaxel?
We conducted a randomized Phase II trial, which we reported in San Antonio last December (Gradishar 2006b) and updated at ASCO this year (Gradishar 2007).
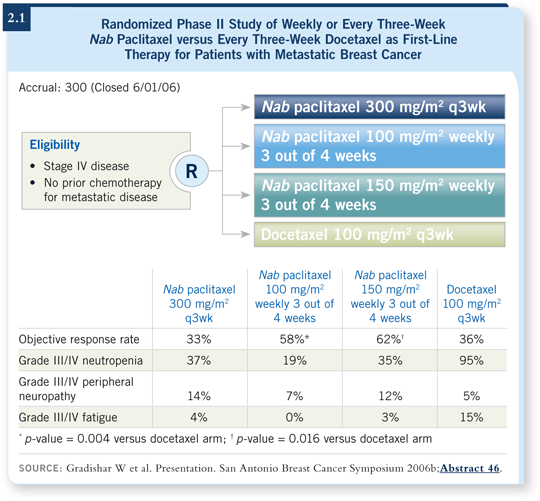
Patients with metastatic breast cancer were randomly assigned to first-line treatment with a dose of 300 mg/m2 of nab paclitaxel every three weeks, 100 mg/m2 of docetaxel every three weeks or nab paclitaxel administered weekly three out of four weeks at a dose of either 100 or 150 mg/m2 (2.1).
In December, we reported that the weekly nab paclitaxel schedules were more active from the standpoint of antitumor activity than either every three-week docetaxel or every three-week nab paclitaxel (Gradishar 2006b).
The weekly treatment arms were not only active but also well tolerated, particularly the 100 mg/m2 dose, which appeared at the time to be the optimal schedule.
The weekly schedule with 150 mg/m2 had a slightly higher response rate, but it also is associated with slightly more toxicity. We did not see much of a difference in terms of progression-free survival between these two arms.
Track 2
![]() DR LOVE: Could you comment on the difference in the efficacy findings you reported at ASCO 2007 compared to San Antonio in 2006?
DR LOVE: Could you comment on the difference in the efficacy findings you reported at ASCO 2007 compared to San Antonio in 2006?
![]() DR GRADISHAR: In December we found that the weekly treatment arms were associated with response rates in the 60-plus percent range, markedly higher than the every three-week treatment arms of either nab paclitaxel or docetaxel (Gradishar 2006b).
DR GRADISHAR: In December we found that the weekly treatment arms were associated with response rates in the 60-plus percent range, markedly higher than the every three-week treatment arms of either nab paclitaxel or docetaxel (Gradishar 2006b).
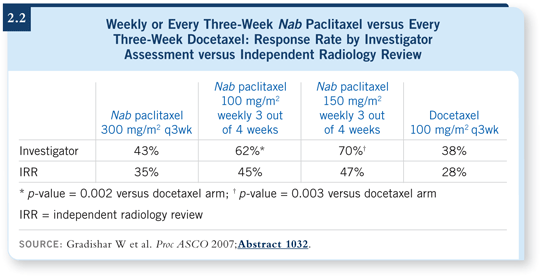
Part of the more recent ASCO presentation was the response rate findings from the independent radiology review (2.2). As expected, there was a drop-off in response rates among all four treatment arms. However, consistent with the original investigator-reported findings, response rates for both weekly nab schedules remained numerically superior to every three-week docetaxel or every three-week nab paclitaxel.
When you include an independent radiology review, you’ll often find that the response rates are less than what the clinicians report. Part of that is because the radiologists are blinded and not directly involved in the trial, so they don’t know what the index lesions are, for instance.
![]() DR LOVE: What was seen with progression-free survival?
DR LOVE: What was seen with progression-free survival?
![]() DR GRADISHAR: Last December we would have said that the progression-free survival is not different across the nab paclitaxel treatment arms, but all are superior to docetaxel administered every three weeks.
DR GRADISHAR: Last December we would have said that the progression-free survival is not different across the nab paclitaxel treatment arms, but all are superior to docetaxel administered every three weeks.
What’s emerging now is that both the every three-week nab paclitaxel and the weekly schedule of 150 mg/m2 nab paclitaxel arms appear to be the superior treatments. But, from the standpoint of efficacy and tolerability, the 150 mg/m2 schedule appears to be the treatment arm to be pursued in a pivotal Phase III trial.
I believe one of the things that will come out of the upcoming randomized trial is whether the added antitumor efficacy that’s presumed to be associated with the weekly schedule will offset what might be slightly more toxicity than we see with lower-dose weekly schedules of nab paclitaxel.
Track 4
![]() DR LOVE: Were there differences in tolerability between the docetaxel and nab paclitaxel arms in your study?
DR LOVE: Were there differences in tolerability between the docetaxel and nab paclitaxel arms in your study?
![]() DR GRADISHAR: One of the interesting observations made across all the reported nab paclitaxel trials is the notion that the neuropathy might be different. One of the first things people would have considered is that with this agent, when you eliminate the Cremophor, no neuropathy should occur.
DR GRADISHAR: One of the interesting observations made across all the reported nab paclitaxel trials is the notion that the neuropathy might be different. One of the first things people would have considered is that with this agent, when you eliminate the Cremophor, no neuropathy should occur.
But what has been observed in every trial — even in the Phase I trials — is that with high doses, you see neuropathy even in the absence of Cremophor. This might be attributable to the chemotherapy drug itself. So neuropathy occurs with nab paclitaxel — that seems to be a consistent finding.
The numbers are not huge, but there appears to be resolution of the neuropathy to the point at which you can readminister the chemotherapy drug within about three weeks.
In other words, you get a decrease in the severity of the neuropathy to the point at which you feel comfortable readministering the drug. That’s in contrast to what we typically see when patients develop Grade III neuropathy with solvent-based paclitaxel, with which the duration of the neuropathy is much longer.
In terms of other side effects, the degree and frequency of significant neutropenia are decreased with the nab paclitaxel every three-week and weekly schedules, relative to the three-weekly docetaxel, and minimal febrile neutropenia is associated with nab paclitaxel at the doses evaluated.
Additionally, in contrast to docetaxel, in our study the incidence of stomatitis is clearly less frequent whether you’re using every three-week or weekly schedules of nab paclitaxel.
Tracks 6-7
![]() DR LOVE: Can you describe the nab paclitaxel Phase III trials currently in development?
DR LOVE: Can you describe the nab paclitaxel Phase III trials currently in development?
![]() DR GRADISHAR: One randomized trial will evaluate patients with metastatic breast cancer receiving therapy with either docetaxel at 100 mg/m2 every three weeks or nab paclitaxel with a 150-mg/m2 weekly schedule.
DR GRADISHAR: One randomized trial will evaluate patients with metastatic breast cancer receiving therapy with either docetaxel at 100 mg/m2 every three weeks or nab paclitaxel with a 150-mg/m2 weekly schedule.
A third arm is building on data Linda Vahdat presented on ixabepilone, which is not a taxane per se but an epothilone (Vahdat 2007). All three arms will evaluate weekly schedules of something that works at the microtubular level.
A separate CALGB study will address the question of weekly schedules of solvent-based paclitaxel compared to nab paclitaxel.
![]() DR LOVE: Is this trial going to include bevacizumab?
DR LOVE: Is this trial going to include bevacizumab?
![]() DR GRADISHAR: It’s not going to include bevacizumab to my knowledge, but some small trials have combined nab paclitaxel with bevacizumab.
DR GRADISHAR: It’s not going to include bevacizumab to my knowledge, but some small trials have combined nab paclitaxel with bevacizumab.
I believe at this point it’s feasible. There have not been any unexpected side effects. More interest will be seen in evaluating that combination moving forward.
![]() DR LOVE: What about the use of nab paclitaxel/bevacizumab in a clinical setting, off protocol?
DR LOVE: What about the use of nab paclitaxel/bevacizumab in a clinical setting, off protocol?
![]() DR GRADISHAR: We have combined nab paclitaxel with bevacizumab. As we see patients with newly diagnosed metastatic breast cancer and try to identify the optimal treatment approach, we are acutely aware of ECOG-E2100, which examined weekly solvent-based paclitaxel with or without bevacizumab (Miller 2005).
DR GRADISHAR: We have combined nab paclitaxel with bevacizumab. As we see patients with newly diagnosed metastatic breast cancer and try to identify the optimal treatment approach, we are acutely aware of ECOG-E2100, which examined weekly solvent-based paclitaxel with or without bevacizumab (Miller 2005).
In ECOG-E2100, the addition of bevacizumab was associated with an enhanced response rate and better progression-free survival. It naturally leads to the question of whether there would be an advantage to using nab paclitaxel, as opposed to solvent-based paclitaxel, in that combination. I believe the simple answer is yes.
There is the sense that nab paclitaxel would be better tolerated than solvent-based paclitaxel over time, with the added benefit that you don’t have to administer steroids, with their associated side effects.
We have administered nab paclitaxel with bevacizumab to patients. I believe an economic issue exists with that, but it’s a reasonable consideration. The drugs are active, and they’re well tolerated.
Track 10
![]() DR LOVE: Some pilot studies evaluated dose-dense AC
DR LOVE: Some pilot studies evaluated dose-dense AC![]() nab paclitaxel. Can you talk about what those have shown and also where you see nab paclitaxel heading in terms of adjuvant therapy?
nab paclitaxel. Can you talk about what those have shown and also where you see nab paclitaxel heading in terms of adjuvant therapy?
![]() DR GRADISHAR: Reports of adjuvant AC
DR GRADISHAR: Reports of adjuvant AC![]() nab paclitaxel demonstrate that you can administer this regimen in a dose-dense fashion. You can administer the every three-week dose on a two-week schedule. So feasibility has been demonstrated (Burstein 2007).
nab paclitaxel demonstrate that you can administer this regimen in a dose-dense fashion. You can administer the every three-week dose on a two-week schedule. So feasibility has been demonstrated (Burstein 2007).
What we don’t know is how it compares directly to other regularly used regimens, such as TAC or dose-dense AC![]() solvent-based paclitaxel. I don’t believe you’ll find some huge surprise substituting nab paclitaxel for any position that a standard taxane holds in the adjuvant setting.
solvent-based paclitaxel. I don’t believe you’ll find some huge surprise substituting nab paclitaxel for any position that a standard taxane holds in the adjuvant setting.
In fact, it is possible that by being able to administer nab paclitaxel at a higher dose safely — in essence, being able to administer more of the taxane safely — you would have greater efficacy.
The only way of addressing that is an enormous randomized trial with thousands of patients. I’m not sure anyone will have the willpower or the resources to do a trial that requires that many patients for that kind of question. So it’s a dilemma.
Another question could be, would you consider using nab paclitaxel in the adjuvant setting as a substitute for a solvent-based paclitaxel? The answer is yes.
We have done it for some patients, generally the rare patients who are either sensitive to the effects of steroids or have experienced some sort of hypersensitivity reaction.
From what we know in the metastatic disease setting, I have absolutely no reason to think nab paclitaxel would not be a good substitute for paclitaxel in the adjuvant setting.
Track 15
![]() DR LOVE: Can you talk about the EFECT study you headed, which evaluated fulvestrant versus exemestane in advanced disease?
DR LOVE: Can you talk about the EFECT study you headed, which evaluated fulvestrant versus exemestane in advanced disease?
![]() DR GRADISHAR: EFECT was an effort to address the issue of what to do for patients who receive nonsteroidal aromatase inhibitors as treatment in the adjuvant or metastatic setting and then develop progressive disease (Gradishar 2006c). This topic has received no shortage of discussion.
DR GRADISHAR: EFECT was an effort to address the issue of what to do for patients who receive nonsteroidal aromatase inhibitors as treatment in the adjuvant or metastatic setting and then develop progressive disease (Gradishar 2006c). This topic has received no shortage of discussion.
Is there an optimal sequence with endocrine therapy? With smaller pilot experiences, we know that you could treat with a different subclass of aromatase inhibitor — in other words, go from a nonsteroidal to a steroidal — and that some fraction of patients respond.
Other pilot studies suggested that you could go from a nonsteroidal to fulvestrant, a selective estrogen receptor regulator, and obtain a clinical response. EFECT attempted to rigorously address the issues of which agent to employ in this setting and whether using a loading dose of fulvestrant might lead to a more rapid achievement of steady-state drug levels.
Preclinical experiments long ago suggested that the FDA-approved dose of fulvestrant was probably on the threshold of obtaining antitumor activity. The question was, could you feasibly administer more volume intramuscularly and reach steady-state levels more quickly?
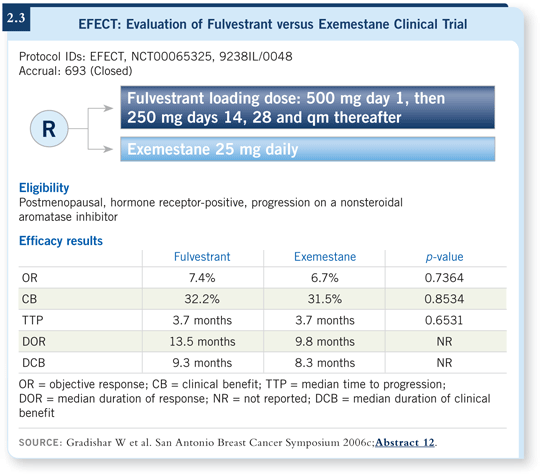
Patients received 500 milligrams on day one — basically a shot in each buttock — and then 250 milligrams on days 14 and 28 and monthly thereafter. Modeling experiments suggested that if you use a higher dose early, you reach the steady state quicker.
A corollary of that is that if you don’t get there through routine dosing, perhaps you might be taking patients off the agent prematurely because they’re simply not experiencing the effect of the drug rapidly enough. Pharmacokinetic analysis from this trial did demonstrate that clinical use of the loading dose mirrors what the modeling predicted.
Regarding efficacy, just about every endpoint was superimposable. Whether you compare response rate, time to disease progression, or even tolerability and adverse events, patients who received either fulvestrant or exemestane had identical outcomes in all of those categories (Gradishar 2006c; [2.3]).
The conclusion is that one could legitimately approach a patient who has experienced progression on a nonsteroidal aromatase inhibitor with either one of these agents. It’s not absolutely clear whether there is a superior sequence you must follow.
Moving forward, we see continued interest in exploring the dose used with fulvestrant. Trials are underway to evaluate continuing 500 milligrams beyond the first dose and to examine the effect of using somewhat higher doses within that early time period.
We also see interest in combining endocrine agents to induce a total estrogen blockade. One approach is to combine an aromatase inhibitor — to eliminate the estrogen or drive it down — with fulvestrant, which in a sense eliminates the receptor. Also, in some trials patients whose disease progresses on anastrozole continue the anastrozole, and fulvestrant is added.

![]()
EDITOR'S NOTE
Peto’s curse
Neil Love, MD
- Select publications
INTERVIEWS
George W Sledge Jr, MD
- Select publications
William J Gradishar, MD
- Select publications
Lee S Schwartzberg, MD
- Select publications
FACULTY TUMOR PANEL
- Select publications
Breast Cancer Update:
A CME Audio Series and Activity