
|
||||||||

| Tracks 1-19 | ||||||||||||||||||||||||||||||||||||||||
|
Select Excerpts from the Meeting
Tracks 6-8
![]() DR WINER: We have single-agent versus combination chemotherapy. In terms
of single-agent chemotherapy, given that she received a taxane 15 months
ago, I’m not terribly enthusiastic about using a taxane again. Of the various
single-agent choices, probably capecitabine is the one for which we have the
most data. It has also been specifically evaluated in patients with disease that is
refractory to an anthracycline and a taxane. Vinorelbine or gemcitabine would also be reasonable choices, although I’m not entirely sure of their activity in
this situation.
DR WINER: We have single-agent versus combination chemotherapy. In terms
of single-agent chemotherapy, given that she received a taxane 15 months
ago, I’m not terribly enthusiastic about using a taxane again. Of the various
single-agent choices, probably capecitabine is the one for which we have the
most data. It has also been specifically evaluated in patients with disease that is
refractory to an anthracycline and a taxane. Vinorelbine or gemcitabine would also be reasonable choices, although I’m not entirely sure of their activity in
this situation.
We have a number of combination therapy options. In the study conducted by Joyce O’Shaughnessy that evaluated docetaxel with or without capecitabine for women with metastatic breast cancer, the combination showed improvements in response rate, time to progression and overall survival. The patients were all docetaxel-naïve, and they weren’t crossed over on a regular basis to capecitabine if they had received docetaxel alone (O’Shaughnessy 2002).
The trial comparing paclitaxel with or without gemcitabine also demonstrated a small survival benefit for the combination. It included no crossover, and all the patients were taxane-naïve (Albain 2004). Given the fact that this particular patient had received a taxane just a little more than a year ago, it’s hard to become enthusiastic about either of these particular combinations.
Capecitabine/vinorelbine is a combination regimen that has been used fairly extensively in clinical trials (Ghosn 2006; Nole 2006). My understanding is that it’s used somewhat more frequently in Europe than in the United States, and I’m not aware of any Phase III trials evaluating it. It certainly uses two active drugs that this patient has not previously seen. If I had no other options and a sense that I might not have the opportunity to use a second regimen, this might be a regimen I would consider.
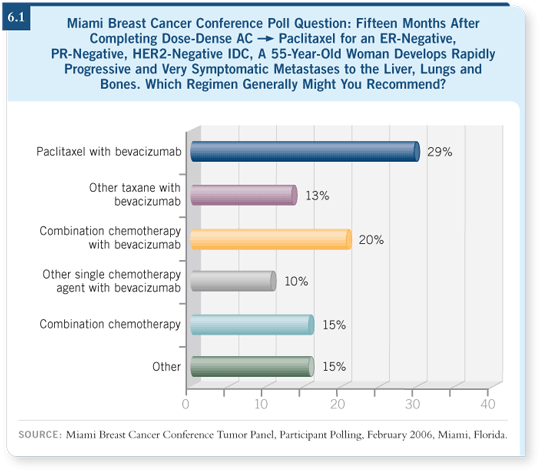
That brings me to bevacizumab and the results from the ECOG-E2100 study, presented by Kathy Miller at ASCO 2005 (Miller 2005a) and the 2005 San Antonio Breast Cancer Symposium (Miller 2005b). ECOG-E2100 included patients who had not received prior chemotherapy for metastatic breast cancer. They were randomly assigned to receive weekly paclitaxel with or without bevacizumab (6.2). Technically, this patient probably would have been eligible for ECOG-E2100 because it excluded patients who had received a taxane within a year but not those who had received a taxane within 15 months.
The addition of bevacizumab to paclitaxel essentially doubled the response rate — the increase was a little more than that if one considers only the patients who had measurable disease. Importantly, progression-free survival showed approximately a five-month improvement, which was highly statistically significant, with the addition of bevacizumab to paclitaxel (Miller 2005b; [6.2]).
In terms of overall survival, when the data were initially presented at ASCO 2005, they suggested a preliminary survival advantage (Miller 2005a). When presented at San Antonio, the advantage was numeric, although it was not considered statistically significant (Miller 2005b). I believe it must be followed over time to determine whether the addition of bevacizumab does, in fact, improve survival.

At the 2005 San Antonio Breast Cancer Symposium, the ECOG investigators presented the benefit in progression-free survival with bevacizumab across different subgroups. It appears that bevacizumab was active in virtually all subgroups. If you specifically consider the patients who had ER-negative and PR-negative disease, you see a clear improvement in progression-free survival within that subgroup (Miller 2005b).
It’s important to keep in mind the prior study of bevacizumab for women with breast cancer. That trial was also conducted by Kathy Miller and evaluated capecitabine with or without bevacizumab. That trial demonstrated a small improvement in response rate but no improvement in progression-free or overall survival (Miller 2005c).
One might ask why those results were so different from the ones from ECOG-E2100. It’s possible this was a chance finding, although I believe that’s unlikely. It’s possible that it’s the use of paclitaxel instead of capecitabine. Preclinical work suggests synergy between the taxanes and bevacizumab. However, in colorectal cancer, bevacizumab has been combined successfully with 5-FU.
Importantly, there were differences in the patient populations. In the capecitabine trial, the majority of the patients had received prior chemotherapy for metastatic disease. Almost all the patients had received some form of prior chemotherapy and an anthracycline and taxane. The study populations were also somewhat different in terms of HER2 status and whether they had received trastuzumab (Miller 2005b, 2005c).
This patient falls somewhere in between those enrolled in ECOG-E2100 and the group of patients in the capecitabine trial. I personally would consider bevacizumab, but I’m not enthusiastic about using it with paclitaxel, despite the fact that that was the regimen used in ECOG-E2100 and she would have been eligible for the trial.
Other options I would tend to consider include bevacizumab with vinorelbine or, perhaps, a taxane/platinum combination. Given the negative trial with capecitabine, I’d probably take that off the list.
We conducted a study led by Hal Burstein evaluating bevacizumab and vinorelbine in 56 patients, which demonstrated a modest degree of activity and an acceptable side-effect profile (Burstein 2002). For this patient, I probably would use bevacizumab with some other chemotherapy drug. I fully recognize that others might want to be purists and say, “This patient was eligible for ECOG-E2100, and I’m going to use paclitaxel.” I don’t believe that is wrong.
If I expected an opportunity for a second agent and I were not going to use bevacizumab, I’d use single-agent capecitabine. Finally, if I felt compelled to use combination therapy — and this may be a situation in which, particularly if you choose not to use bevacizumab, you might choose to do that, given the extent of the patient’s disease and her symptoms — I would use a two-drug regimen with drugs she’s never received before.
Track 14
![]() DR ROBERTSON: We have no randomized, controlled data on endocrine
therapies for metastatic breast cancer following an adjuvant aromatase inhibitor.
In fact, we have very little Phase II data. So we’re going to have to take
data from other situations and apply them.
DR ROBERTSON: We have no randomized, controlled data on endocrine
therapies for metastatic breast cancer following an adjuvant aromatase inhibitor.
In fact, we have very little Phase II data. So we’re going to have to take
data from other situations and apply them.
Exemestane, a steroidal aromatase inhibitor, shows approximately a 30 percent clinical benefit rate following a nonsteroidal aromatase inhibitor (Carlini 2002). Fulvestrant shows a similar clinical benefit rate of around 30 percent following an aromatase inhibitor (Perey 2004). The numbers in these Phase II sequence studies, however, are pretty small, and the hormonal therapy was used as third-line therapy.
Another option would be to continue the anastrozole and start fulvestrant. The basis for that is preclinical data that were published in The Journal of Biological Chemistry by Dr Martin (Martin 2003). If you grow cells in estrogen-deprived media, they become more sensitive to low levels of estrogen. People assume that perhaps, in patients who’ve been treated with an aromatase inhibitor over a long period of time, this is what happens. It’s one of the potential mechanisms of resistance to aromatase inhibitors.
If you take cells that have been estrogen deprived for a long time and administer increasing doses of fulvestrant, then you can inhibit cell growth. The hypothesis is that when you have relapse on an aromatase inhibitor and the tumor becomes sensitive to these very low levels of estradiol, if you simply use fulvestrant and stop the aromatase inhibitor, then you increase estradiol again, which might compete with fulvestrant. Therefore, perhaps it may be better to keep the estradiol level low and bring in fulvestrant.
It’s a great theory, which is being tested at the moment in an ongoing trial in the United Kingdom called the SoFEA study (6.4). We have no data, and we’re probably not going to have a lot of clinical data in the next year or two.
The other option is tamoxifen. Again, we have no good randomized data. We have Phase II data from a first-line study of an aromatase inhibitor versus tamoxifen. From the patients who received the aromatase inhibitor, data were collected for those who went on to receive tamoxifen. A clinical benefit rate of approximately 50 percent appeared among patients with advanced breast cancer who received tamoxifen as second-line therapy following an aromatase inhibitor (Thürlimann 2004).
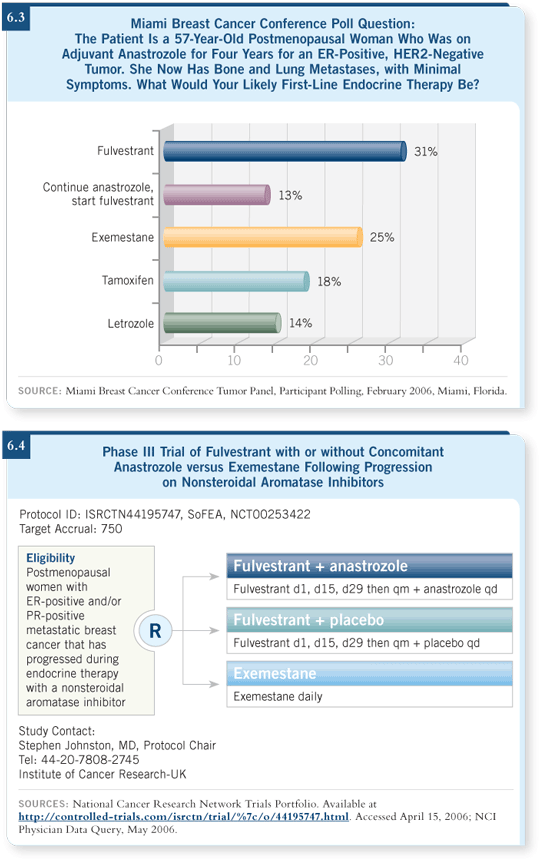
I personally would like to use a different mechanism of action, and I’d use an antiestrogen. I believe the clinical benefit rate with tamoxifen following an aromatase inhibitor as first-line therapy is high. Also, this tumor is HER2-negative.
If you believe breast cancer is less responsive if it’s HER2-positive, then that would be another reason to be comfortable with tamoxifen. If I weren’t going to use tamoxifen, then I would select another antiestrogen. My second choice would be fulvestrant.
Tracks 15, 16
![]() DR ROBERTSON: It takes about three to four months to reach a steady state
with fulvestrant. However, if you administer 250 mg on days one, 14 and 28,
and then repeat it every 28 days, you achieve a steady state much faster.
The question is, should you administer an extra dose at day 14? I have to say,
“Not usually.” I have used it for certain patients when I may have one chance
at endocrine therapy.
DR ROBERTSON: It takes about three to four months to reach a steady state
with fulvestrant. However, if you administer 250 mg on days one, 14 and 28,
and then repeat it every 28 days, you achieve a steady state much faster.
The question is, should you administer an extra dose at day 14? I have to say,
“Not usually.” I have used it for certain patients when I may have one chance
at endocrine therapy.
If you’re coming back to fulvestrant after chemotherapy, usually this is for patients with visceral disease. Even then, it’s a small minority of cases. If they have nothing left to try, and they have ER-positive disease, I would use a loading dose. Normally, I would use 250 mg every 28 days.
![]() DR LOVE: Kent, do you utilize that strategy?
DR LOVE: Kent, do you utilize that strategy?
![]() DR OSBORNE: Yes, I have with some patients — the issue is how fast you
need to reach therapeutic levels. I may take that approach in a patient who has
more aggressive disease and the therapeutic levels need to be reached faster.
However, in a patient with bone-only indolent disease, I’d probably utilize the
once-a-month schedule.
DR OSBORNE: Yes, I have with some patients — the issue is how fast you
need to reach therapeutic levels. I may take that approach in a patient who has
more aggressive disease and the therapeutic levels need to be reached faster.
However, in a patient with bone-only indolent disease, I’d probably utilize the
once-a-month schedule.
Tracks 17, 18
![]() DR SMITH: We don’t really have an algorithm. I would be inclined to use
fulvestrant with estrogen deprivation. In other words, I would continue an
aromatase inhibitor because I can’t see any reason why it would be worse than
fulvestrant alone. Until the trial results are reported, that would be the option
I’d choose.
DR SMITH: We don’t really have an algorithm. I would be inclined to use
fulvestrant with estrogen deprivation. In other words, I would continue an
aromatase inhibitor because I can’t see any reason why it would be worse than
fulvestrant alone. Until the trial results are reported, that would be the option
I’d choose.
![]() DR LOVE: Dan, can you talk about how you decide between fulvestrant,
tamoxifen, and anastrozole for postmenopausal patients?
DR LOVE: Dan, can you talk about how you decide between fulvestrant,
tamoxifen, and anastrozole for postmenopausal patients?
![]() DR HAYES: With my own patients, I say, “Would you rather have an injection
or take a pill?” If they’d rather have an injection, then I suggest fulvestrant. I
do use the loading dose.
DR HAYES: With my own patients, I say, “Would you rather have an injection
or take a pill?” If they’d rather have an injection, then I suggest fulvestrant. I
do use the loading dose.
If they would rather take a pill, I suggest tamoxifen. I believe there’s a reason, theoretically, that either one of them would be likely to work.
Surprisingly, in my practice it’s 50-50. I always guess wrong what people will choose to do. So it’s nice to have a couple of options.

![]()
Editor’s Note:
Reality check
Interviews
Marc E Lippman, MD
- Select publications
C Kent Osborne, MD
- Select publications
I Craig Henderson, MD
- Select publications
Charles E Geyer Jr, MD
- Select publications
Frank A Vicini, MD
- Select publications
Miami Beach Cancer Conference Tumor Panel Discussion on Systemic Therapy of Metastatic Disease
- Select publications